Proyecto MACH
Información básica para acceder al servidor Soroban, comandos iniciales para cargar librerias/programas y primeros pasos para utilizar el gestor de recursos SLURM se presenta a continuación.
Acceso remoto a Soroban
- Solicitar una cuenta de acceso a andres.avila@ufrontera.cl
- Un cliente ssh instalado en tu computador personal
- Conocimientos básicos de Linux y su linea de comando (CLI)
Aplicaciones disponibles en Soroban (system)
Compiladores, interpretes, librerias, y utilidades
índice : A|B|C|D|E|F|G|H|I|J|K|L|M|N|O|P|Q|R|S|T|U|V|W|X|Y|Z
| Software | Versión | Descripción | |
|---|---|---|---|
| A | |||
| Anaconda python | 3.7.8 | Interprete python optimizado | |
| B | |||
| C | cmake | 3.15.4 | Utilidad para compilar en Linux |
| D | |||
| E | |||
| F | |||
| G | |||
| GCC | 8.3.1 | Compilador c/c++/fortran | |
| GCC | 5.5.5 | Compilador c/c++/fortran | |
| Go | 1.15.2 | Compilador de Lenguaje GO | |
| Clang | 3.4.2 | Compilador c/c++ de LLVM | |
| H | |||
| HDF5 | 1.10.5 | FileSystem Distribuido | |
| I | |||
| J | |||
| Java Develop Kit (JDK) | 11.0.8 | JDK (openjdk) librerias/compilador JAVA | |
| K | |||
| L | |||
| M | |||
| Math Kernel Library (MKL) | 2018.4-274 | Librerias matemáticas optimizadas de Intel | |
| N | |||
| O | |||
| openmpi | 1.10.7 | Programación paralela | |
| openmpi | 3.1.5 | Programación paralela | |
| openmpi | 1.6.5 | Programación paralela | |
| openmpi | 2.1.6 | Programación paralela | |
| openblas | 3.3-2 | Implementación opensource de BLAS y LAPACK ; algebra lineal | |
| P | |||
| Perl | 5.16.3 | Interprete del lenguaje Perl | |
| Q | |||
| R | |||
| S | |||
| T | |||
| U | |||
| V | |||
| W | |||
| X | |||
| Y | |||
| Z |
Aplicaciones Científicas
Bioinformatics, Chemistry, Physics, etc
índice : A|B|C|D|E|F|G|H|I|J|K|L|M|N|O|P|Q|R|S|T|U|W|X|Y|Z
| # | Aplicación | Versión | Descripción |
| A | |||
| aragorn | 1.2.38 | tRNA (and tmRNA) detection | |
| arb | 6.0.6 | Phylogenetic analysis of rRNA and other biological sequences such as amino acids | |
| ascp | 3.9.9 | – | |
| B | |||
| bowtie2 | 0.7.17 | software package for mapping low-divergent sequences against a large reference genome, such as the human genome | |
| bwa | 2.4.1 | alineación de secuencias y el análisis de secuencias en bioinformática | |
| blast+ | 2.9.0 | Alineamiento de secuencias de tipo local, ya sea de ADN, ARN o de proteínas | |
| blast+ | 2.10.0(default) | Alineamiento de secuencias de tipo local, ya sea de ADN, ARN o de proteínas | |
| blast | 2.2.26 | Alineamiento de secuencias de tipo local, ya sea de ADN, ARN o de proteínas | |
| bedtools | 2.29.2 | swiss-army knife of tools for a wide-range of genomics analysis tasks | |
| bedops | 2.4.38 | highly scalable and easily-parallelizable genome analysis toolkit | |
| beagle-lib | 3.1.2 | high-performance library that can perform the core calculations at the heart of most Bayesian and Maximum Likelihood phylogenetics | |
| bcftools | 1.10.2 | set of utilities that manipulate variant calls | |
| bbtools | 38.86 | suite of fast, multithreaded bioinformatics tools designed for analysis of DNA and RNA sequence data | |
| barrnap | 0.9 | predicts the location of ribosomal RNA genes in genomes | |
| bamtools | 2.5.1 | toolkit for handling BAM files. | |
| C | |||
| cufflinks | 2.10 | finds and removes adapter sequences, primers, poly-A tails and other types of unwanted sequence from your high-throughput sequencing reads | |
| circos | 2.2.1 | Transcriptome assembly and differential expression analysis for RNA-Seq. | |
| cutadapt | 0.69-9 | for visualizing data and information. It visualizes data in a circular layout | |
| checkm | 1.1.2 | set of tools for assessing the quality of genomes recovered from isolates, single cells, or metagenomes | |
| cdhit | 4.8.1 | Takes a fasta format sequence database as input and produces a set of 'non-redundant' (nr)… | |
| canu | 1.9 | Fork of the Celera Assembler designed for high-noise single-molecule sequencing | |
| D | |||
| diamond | 0.9.35 | molecular and crystal structure visualization software | |
| E | |||
| F | |||
| G | |||
| gromacs | 2019.4 | Modelamiento Molecular | |
| H | |||
| hmmer | 2.3.2 | biosequence analysis using profile hidden Markov models . | |
| hmmer | 3.1b1 | biosequence analysis using profile hidden Markov models . | |
| hmmer | 3.1b2 | biosequence analysis using profile hidden Markov models . | |
| hmmer | 3.2.1 | biosequence analysis using profile hidden Markov models . | |
| I | |||
| J | |||
| K | |||
| L | |||
| M | |||
| N | |||
| Namd2 | 2019-11-27-multicore | Modelamiento Molecular | |
| P | |||
| Q | |||
| R | |||
| R | 3.6.1 | Analísis Estadistico | |
| W | |||
| WRF | 3.9.1 | Mesoscale numerical weather prediction | |
| WRF | 4.0 | Mesoscale numerical weather prediction | |
| X | |||
| 6 | netcdf/c | 4.7.3 | Network Common Data Form |
| 7 | netcdf/fortran | 4.5.2 | Network Common Data Form |
| 8 | quantum-espresso/schrodinger | 6.4.1-2019-4 | Modelamiento Molecular |
| 32 | eggnog-mapper | 2.0.1 | tool for functional annotation of large sets of sequences based on fast orthology assignments using precomputed clusters |
| 33 | EMBOSS | 6.6.0 | European Molecular Biology Open Software Suite |
| 34 | FastANI | 1.3 | Produces accurate ANI estimates and is faster than alignment (e.g., BLAST)- based approaches. |
| 35 | vcftools | 0.1.17 | package designed for working with VCF files |
| 36 | Unicycler | 0.4.9b | assembly pipeline for bacterial genomes |
| 37 | tRNAscan-SE | 2.0.5 | searches for tRNA genes in genomic sequences |
| 38 | trinityrnaseq | 2.11.0 | Trinity RNA-Seq de novo transcriptome assembly |
| 39 | trf | 4.09 | Analyze DNA sequences |
| 40 | TransDecoder | 5.5.0 | Assist in the identification of potential coding regions within …. |
| 41 | tophat | 2.1.1 | Alineación del rendimiento de las lecturas de secuenciación de cDNA … |
| 42 | tbl2asn | 3.10.0 | Automates the creation of sequence records for submission to GenBank |
| 43 | ssu-align | 0.1.1 | identifying, aligning, masking and visualizing archaeal 16S, bacterial 16S and eukaryotic 18S… |
| 44 | sratoolkit | 2.10.7 | Using the SRA Toolkit to convert .sra files into other formats … |
| 45 | SPAdes | 3.14.1 | Assembly toolkit containing various assembly pipelines. |
| 46 | SPAdes | 3.13.2 | Assembly toolkit containing various assembly pipelines. |
| 47 | SINA | 1.6.1 | accurate high-throughput multiple sequence alignment of ribosomal RNA genes. |
| 48 | seqtk | 1.3 | A fast and lightweight tool for processing sequences in the FASTA or FASTQ format. |
| 49 | seqkit | 0.12.1 | Seqkit is a tool for manipulating fasta and fastq files. |
| 50 | samtools | 1.10.2 | suite of programs for interacting with high-throughput sequencing data |
| 51 | salmon | 1.2.1 | Is a tool for quantifying the expression of transcripts using RNA-seq data. |
| 52 | RSEM | 1.3.3 | Tool for the quantification of RNA-seq data. |
| 53 | roary | 1.7.7 | Application for rapidly constructing pan genomes from large numbers of prokaryote sample |
| 54 | racon | 1.4.16 | Consensus sequence |
| 55 | quast | 5.1.0rc1 | Quality Assessment Tool for Genome Assemblies |
| 56 | pyani | 0.2.10 | a Python3 module that provides support for calculating average nucleotide identity (ANI) |
| 57 | prokka | 1.14.6 | tool for the rapid annotation of prokaryotic genomes |
| 58 | prodigal | 2.6.3 | Prodigal is an unsupervised machine learning algorithm |
| 59 | prinseq | 0.20.4 | Tool that generates summary statistics of sequence and quality data and that is used to filter, reformat and trim … |
| 60 | prank | 170427 | Is a probabilistic multiple alignment program for DNA, |
| 61 | pplacer | 1.1.alpha17 | Places query sequences on a fixed reference phylogenetic tree to maximize phylogenetic likelihood or posterior probability according to a reference … |
| 62 | htslib | 1.10.2 | An implementation of a unified C library for accessing common file formats, such as SAM, CRAM and VCF, used for high-throughput sequencing data, … |
| 63 | mash | 2.2 | Reduces large sequences and sequence sets to small, … |
| 64 | mafft | 7.470 | is a multiple sequence alignment program for unix-like operating systems |
| 65 | ksnp | 3.1.2 | Finds single nucleotide polymorphisms (SNPs) in whole genome data. |
| 66 | krona | 2.7.1 | powerful metagenomic visualization tool |
| 67 | kaiju | 1.7.3 | program for the taxonomic classification of high-throughput sequencing reads. |
| 68 | jellyfish | 2.3.0 | tool for fast, memory-efficient counting of k-mers in DNA. |
| 69 | jdk | 8.0_41 | Java Develop ToolKit |
| 70 | jdk | 14.0.1 | Java Develop ToolKit |
| 71 | jdk | 11.28 | Java Develop ToolKit |
| 72 | infernal | 1.1.3 | for searching DNA sequence databases for RNA structure and sequence similarities |
| 73 | pilon | 1.23 | an automated genome assembly improvement and variant detection tool |
| 74 | picard | 2.23.1 | a set of command line tools for manipulating high-throughput sequencing (HTS) data and formats such as SAM/BAM/CRAM and VCF. |
| 75 | phyml | 3.3.20190909 | is a phylogeny software based on the maximum-likelihood principle |
| 76 | phylip | 3.697 | a free package of programs for inferring phylogenies |
| 77 | parallel | 20200622 | Parallel is an indispensible tool for speeding up bioinformatics. It allows you to easily parallelize commands |
| 78 | paml | 4.9 | a package of programs for phylogenetic analyses of DNA or protein sequences using maximum likelihood. |
| 79 | ncftp | 3.2.6 | cliente ftp |
| 80 | muscle | 3.8.31 | A program to create multiple sequence alignments of a large number of sequences |
| 81 | MUMmer | 3.23 | an open source software package for the rapid alignment of very large DNA and amino acid sequences |
| 82 | mothur | 1.44.1 | a single resource to analyze molecular data that is used by microbial ecologists |
| 83 | minimap2 | 2.17 | pairwise alignment for nucleotide sequences. |
| 84 | miniconda | 4.8.3 | python minimalist |
| 85 | MCL | 14-137 | a cluster algorithm for graphs |
| 86 | MinCED | 0.4.2 | program to find Clustered Regularly Interspaced Short . |
| 87 | fastqc | 0.11.9 | a quality control application for high throughput sequence data. |
| 88 | fasttree | 2.1.11 | FastTree infers approximately-maximum-likelihood phylogenetic trees from alignments of nucleotide or protein sequences |
| 89 | Gblocks | 0.91b | computer program written in ANSI C language that eliminates poorly aligned positions and divergent regions of an alignment of DNA or protein |
| 90 | go | 1.14.4 | compilador de lenguaje GO |
| 91 | gtdbtk | 1.2.0 | a toolkit to classify genomes with the Genome Taxonomy Database, B |
Activación de MFA (opcional)
Multi Factor Authenticator (MFA) . Este método agrega un capa de seguridad extra a las cuentas de los investigadores, y es obligatorio para todos los usuarios.
Para activar y usar MFA es necesario seguir tres simples pasos descritos a continuación:
Paso uno

Instalar la apps de google authenticator en el smartphone personal (android , iOS) desde google play o apple store respectivamente.
Paso dos
IMPORTANTE: Puedes saltar las intrucciones 1 y 2, en caso de ya tengas una ssh-key, solamente debes agregarla al servidor Soroban como se indica en la instrucción 3, con tu ya existente ssh-key.
El uso de keypair es un proceso opcional,pero altamente recomendado tanto para usarios Windows, Linux y OSX.
Si no desean crear y gestionar claves públicas y privadas, pueden saltar ese paso,aun asi MFA funcionará solicitando el código de verificación único en el tiempo en cada inicio de session.
Al gestionar las conexiones SSH con claves públicas/privadas la máquina cliente (usuario) estará autorizado y no será necesario otro medio de autenticación, el ingreso será directo, al existir una relación de confianza gestionada por las claves previamente generadas como se muestra en los siguientes pasos, esto tambien aplica al resto de los sistemas operativos (Linux, OSX, FreeBSD, etc).
Aun asi, este proceso puede ser realizado en cualquier momento si asi se desea.
Es necesario crear ssh-key para Linux, OSX o Windows, según sea el caso. SSH-Key facilita el acceso al servidor Soroban o cualquier otro servidor eliminando el ingreso de la password en cada inicio de session ssh.
Para los usuarios Linux y OSX tienen disponible el siguiente método basado en CLI (Command Line Interface) cualquier otro método es completamente válido, pero este es un procedimiento universal disponible en todos las versiones de linux, freebsd, unix, y OSX, para esto debe abrir una terminal y ejecutar las siguientes 3 instrucciones:
Instrucción 1
ssh-keygen -o -a 100 -t ed25519 -f ~/.ssh/id_ed25519-soroban
El comando anterior crea una ssh-key con el algoritmo de cifrado ed25519 y de nombre id_ed252519-soroban, nombre del archivo que puede ser elegido por el usuario.
Instrucción 2
ssh-add .ssh/id_ed25519-soroban
Este comando agrega la ssh-key al llavero de claves para facilitar el uso y especialmente cuando existente en el sistema otras ssh-key para otros servidores.
Instrucción 3
ssh-copy-id -p 10170 -i ~/.ssh/id_ed25519-soroban youruser@paliaike.ingmat.ufro.cl:
Con este comando instala las ssh-key, su parte pública, en el servidor Soroban. Dónde “user” es el usuario en el servidor soroban.
Usuarios Windows (ssh-key)
El uso de keypair es un proceso opcional,pero altamente recomendado tanto para usarios Windows, Linux y OSX.
Si no desean crear y gestionar claves públicas y privadas, pueden saltar ese paso,aun asi MFA funcionará solicitando el código de verificación único en el tiempo en cada inicio de session.
Al gestionar las conexiones SSH con claves públicas/privadas la máquina cliente (usuario) estará autorizado y no será necesario otro medio de autenticación, el ingreso será directo, al existir una relación de confianza gestionada por las claves previamente generadas como se muestra en los siguientes pasos, esto tambien aplica al resto de los sistemas operativos (Linux, OSX, FreeBSD, etc).
Aun asi, este proceso puede ser realizado en cualquier momento si asi se desea.
1. Descargar el cliente putty y puttygen
2. Crear las claves pública y privada (puttygen)
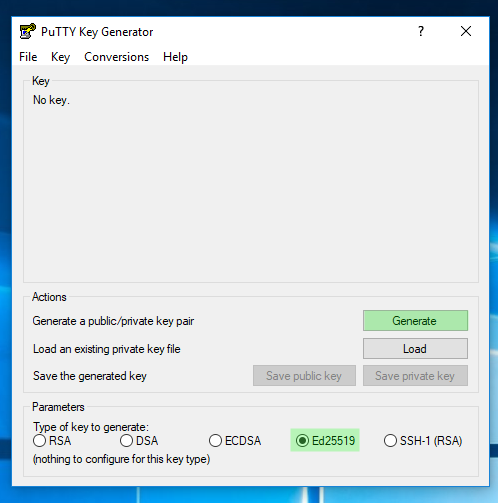
IMAGEN 1: Generar keypair con el algoritmo ED25519 (recomendado)
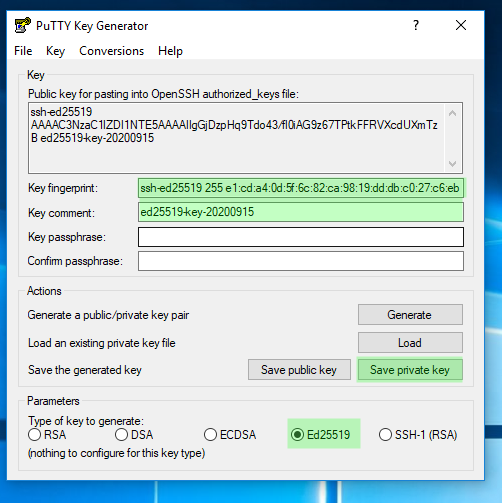
IMAGEN 2: Guardar en un lugar seguro para usarse durante las sessiones SSH hacia el servidor Soroban
3. Copiar la clave pública al servidor Soroban
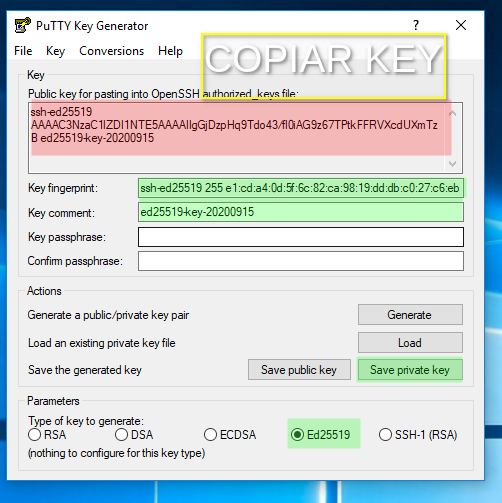
IMAGEN 3 : Copiar clave pública
Copiar la clave pública generada como muestra la imagen, esta puede ser leida desde los archivo creados y guardados como se muestra en la imagen 2 en cualquier momento, esta clavé publica es la que debe ser copiada en el servidor para la validación.
Una vez copiado el texto que representa la clave pública como muestra la imagen 3, debes usar tu cliente ssh para windows, este ejemplo usa putty.
Conectar al servidor Soroban y agregar al final la clave publica editando el siguiente archivo con el siguiente comando:
nano ~/.ssh/authorized_keys
El editor de texto del ejemplo es nano, pueden utilizar el editor que más les agrade o simplifique el trabajo, lo importante es copiar y pegar al final del archivo la clave pública.
Si todo ha funcionando debes desconectarte del servidor Soroban y configurar putty para que utilice tu clave privada regenerada que será el complemento a la parte publica que acabas de registrar en el servidor. Es importante proteger la clave privada.
4. Conectando al servidor Soroban utilizando la clave privada
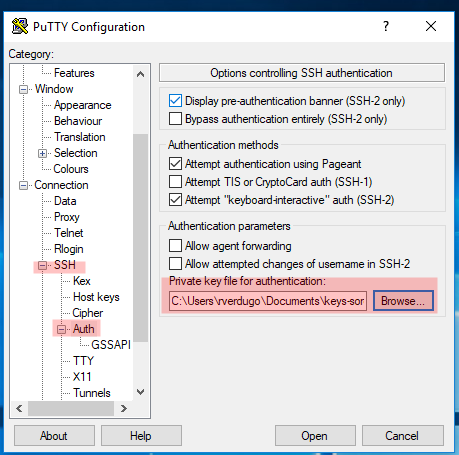
IMAGEN 4: Abierto el cliente putty para windows la imagen resalta en rojo la ubicacion donde debe registrarse la clave privada generada en la IMAGEN 1 y registrada en el servidor Soroban en la IMAGEN 3 bajo el punto 2 “Copiar la clave pública al servidor”.
Esto finaliza el proceso para generar, registrar, configurar y usar keypair para la autentificacion ssh en el servidor Soroban desde un cliente ssh en sistema operativo MS Windows 10, aplicable a cualquier otro servidor.
El proceso varia segun el cliente ssh y la interface de usuario correspondiente, pero en terminos generales son las mismas etapas para cualquiera
Paso tres
Después de completar el paso 1 y paso dos (opcional) debes ingresar al servidor Soroban con tu cliente ssh, independiente del sistema operativo que utilices, y ejecutar en tu cuenta del servidor sorobanel siguiente comando para configurar y activar google authenticator.
Instrucción 1
google-authenticator
Confirmar todas las preguntas con “Y” y guardar en un lugar seguro la secret key y los 5 códigos de emergencia justo abajo del código QR como se muestra en la IMAGEN 1. Las cuales permiten acceso al servidor en caso de tener problemas con el smartphone o cualquier, etc. Importante, cada código puede usarse una única vez.
El código QR debe ser escaneado desde la apps de google authenticator, y con eso queda activo MFA.
Para un proceso manual, en reemplazo del código QR dado que no puedes leer correctamente el QR puedes ingresar directamente en la apps el secret key ubicado justo debajo del código QR para vincular la cuenta en el servidor Soroban y la apps google authenticator. En tu apps verás algo asi “username username-soroban@soroban” que representa la vinculacion de tu cuenta en el servidor soroban con google authenticator.
Resumiendo, ver IMAGEN 1, para completar la activación de MFA tienes dos opciones: una automatica escaneando el código QR y otra manual ingresando directamente el secret key en tu apps del smartphone Android o IOS.

IMAGEN 1: Google Authenticator para MFA
Restauración de Google Authenticator
En situaciones especiales como perdida, daño, desinstalación o simplemente cambio de smartphone estan disponibles varias alternativas:
- Utilizar uno de los 5 códigos de emergencias descritos en el paso 3 para accesar cuando google authenticator no este disponible. Eliminar el archivo .google_authenticator en tu cuenta en Soroban ejecutando el siguiente comando: rm -rf .google_authenticator , y luego repetir el paso 3.-
- - Ingresar el actual secret key directamente en la nueva instalación de google authenticator en el smartphone para volver activar MFA.- -
- Contactar como último recurso al siguiente correo en cualquier horario para soporte : raphael.verdugo@ufrontera.cl
Primeros pasos en Soroban
Cargando un software/libreria en Soroban
Utilización de module
El comando module es utilizado para cargar/descargar software necesario para trabajar; los usuarios pueden acceder de una forma sencilla a cualquier versión de una misma aplicación.
Los usuarios antes de iniciar cualquier cálculo deben cargar las librerias/aplicaciones que utilizarán, siguiendo las siguientes instrucciones:
1. Mostrar librerias y programas disponibles
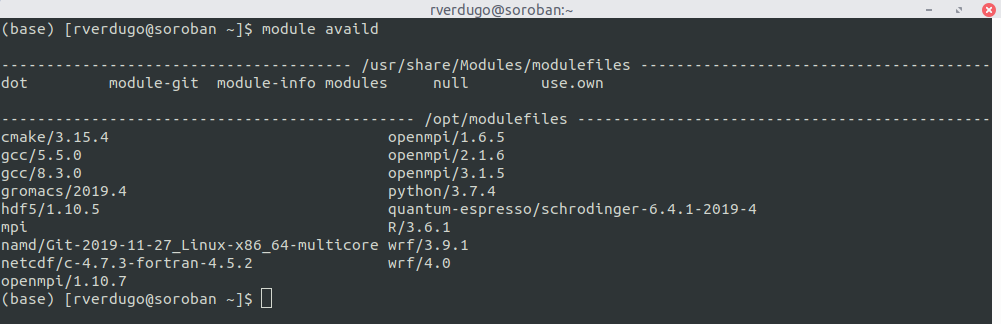
2. Cargar libreria o programa
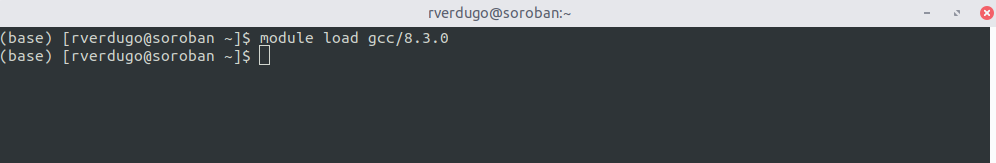
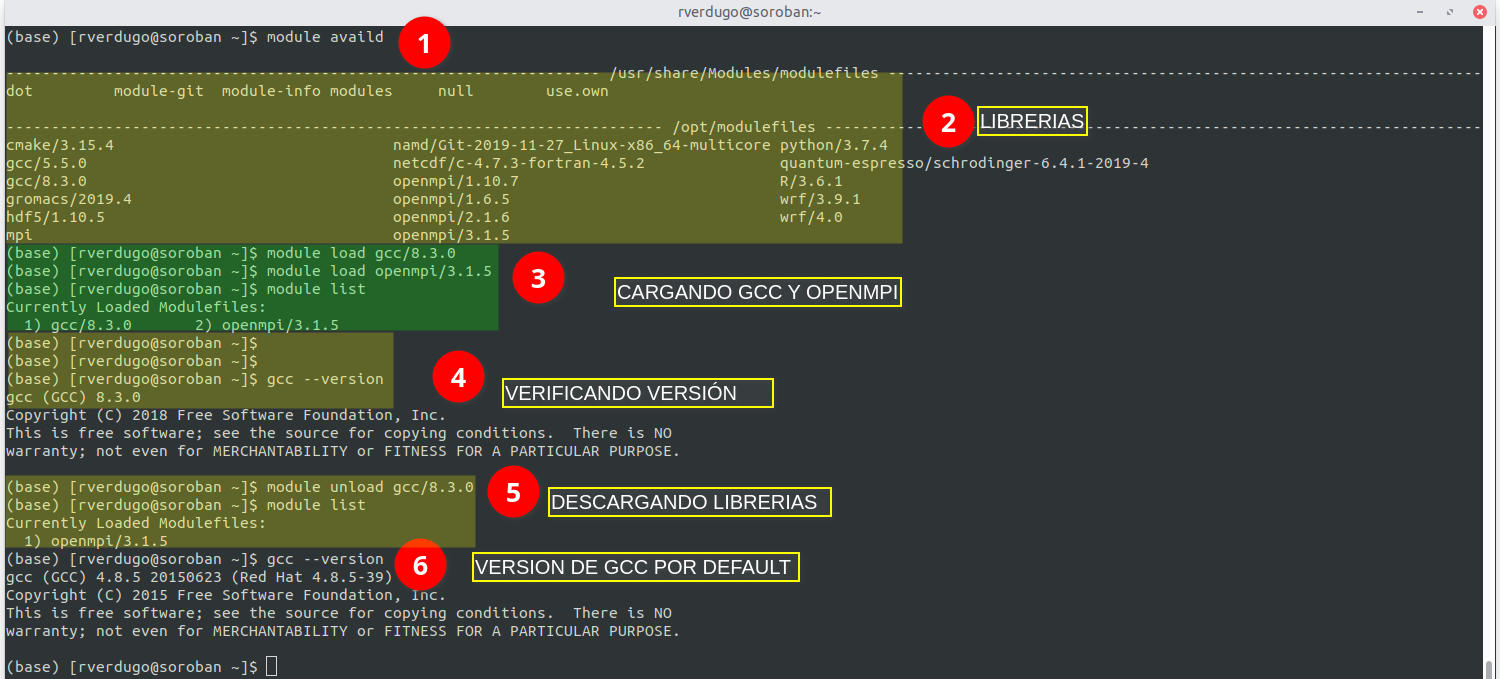
3. Ejemplo Completo
En este ejemplo se muestra el uso del comando module para cargar el compilador gcc 8.3.0 y la libreria openmpi 3.1.5.
El ejemplo contiene los comandos necesarios para :
- Listar las aplicaciones disponibles.
- Cargar los programas/librerias deseados.
- Verificar que este disponible la libreria/aplicación que se ha cargado.
- Descargar las aplicaciones cuando no sean necesarias o se desee otra versión.
Ejecutar un trabajo sin SLURM
Ejecutar una simulación con screen
(multiplexor screen)
Temporalmente es permitido ejecutar simulaciónes sin el encolador SLURM, y el método consiste en estos pasos:
Paso 1
Ejecutar en el servidor lo siguiente:
screen -S nombre-de-tu-trabajo -L
-S : Asigna un nombre a la session llamado “nombre-de-tu-trabajo” , nombre elegido por el usuario
-L : genera en el directorio actual un archivo de nombre screenlog.0 que almacena toda la salida y actividad de la session para su revisión posterior.
Paso 2
python tu-codigo.py
Este ejemplo ejecuta un código python a través del multiplexor screen.
IMPORTANTE: para trabajos de larga duración presiona las teclas: control+a+d . Luego de eso puedes cerrar tu session en el servidor y regresar otro día mientras el servidor continua trabajando.
Paso 3
Para recuperar o restaurar una session anterior debes ejecutar en el servidor lo siguiente:
screen -list #lista todas las sessiones abiertas (detached) que puede ser recuperadas
There is a screen on:
26886.nombre-de-tu trabajo (09/11/20 20:54:43) (Detached)
screen -r 26886.nombre-de-tu trabajo #recuperar una session detached para continuar trabajando en ella
IMPORTANTE: Toda la actividad de la session screen quedará registrada en el archivo de texto screenlog.0 para revisión y analisis de la ejecución de tu trabajo. Cada vez que restaures una session, y el proceso no ha terminado debes repetir los comandos control+a+d para que la session no se pierda, y asi volver en otro momento.
Video Demostrativo
Utilización de SLURM
SLURM Workload Manager o formalmente (Simple Linux Utility for Resource Management) es un sistema de manejo de cluster y calendarización de tareas para grandes y pequeños cluster Linux, el cual es open source, tolerante a fallas y altamente escalable.
SLURM cumple tres funciones principales
- Asigna acceso exclusivo y/o no exclusivo a recursos (nodos de cómputo) a usuarios por un tiempo determinado para que estos puedan ejecutar una tarea. - Provee un framework para iniciar, ejecutar y monitorear tareas (normalmente tareas paralelas) en un conjunto de nodos asignados. - Coordina la solicitud de recursos a través de una cola de tareas pendientes.
¿Cómo se encarga Slurm de manejar los jobs?
- Asignando recurso de cómputo solicitados por el job * Ejecutando el job y * Reportando la salida de la ejecución al usuario.
Ejecutar un job requiere al menos los siguientes pasos:
- Preparar un script * Enviar job para ejecución.
1. Comandos básicos de Slurm
| Comando | Descripción |
|---|---|
| squeue | ver información de jobs en cola |
| sinfo | ver cola, partición e información del nodo |
| sbatch | enviar un job a través de un script |
| srun | enviar un job interactivo |
| scancel | cancelar jobs en cola |
| scontrol | control e información detallada de jobs, colas y particiones. |
| sstat | ver a nivel de sistema la utilización de recursos (memoria, I/O, energía) |
| sacct | ver a nivel de sistema la utilización de recursos de jobs completados. |
2. Ejemplo básico 1 (multi-thread con OpenMP)
Este es un ejemplo de un script (ejemplo1.sh) con los elementos mínimos para ejecutar el programa namd a través de slurm:
#!/bin/bash #Shell a usar durante la simulación para este caso es bash #SBATCH -J NAMD-NOMBRE-SIMULACION #Nombre de la simulacion reemplazar por nombre apropiado #SBATCH --nodes=1 #Numero de nodos donde se ejecutara la simulacion, siempre es 1 para Soroban #SBATCH --tasks-per-node=40 #40 es el numero de procesos a ejecutar en el servidor Soroban, reemplazar por la cantidad adecuada #SBATCH --mem=100G #Memoria reservada para esta simulacion en el servidor #SBATCH --partition=intel #Particion donde se enviaran los trabajos en este caso la particion general se llama intel. module load namd/Git-2019-11-27_Linux-x86_64-multicore #programas o modulos necesarios para ejecutar esta simulacion, reemplazar por las adecuadas en cada caso
Para enviar este script a slurm, crear un job, y comenzar el procesamiento se requiere lo siguiente:
chmod +x ejemplo1.sh
sbatch ejemplo1.sh
3. Ejemplo básico 2 (single thread)
Este es un ejemplo de un script (ejemplo2.sh) con los elementos minimos para ejecutar el programa R-3.6.1 a través de slurm:
#!/bin/bash #shell usada por el usuario, para este baso es BASH #SBATCH -J R-NOMBRE-SIMULACION #Reemplazar R-nombre-simulacion por el nombre correspondiente a la simulación #SBATCH --nodes=1 #Número de nodos a donde se enviara la simulacion, para soroban es 1. #SBATCH --tasks-per-node=1 #Numero de tares por nodo, en este caso es 1 porque es una tarea single-thread #SBATCH --mem=100G #cantidad de memoria reservada para esta simulacion, 100G, reemplazar por valor adecuado #SBATCH --partition=intel #Nombre de la particion slurm donde se enviara las simulaciones en este caso la particion general se llama "intel" module load R/3.6.1 "Modulos necesarios para ejecutar la simulacion, para este ejemplo solamente es necesario 'module load R/3.6.1' este ejemplo es para single thread. <font 16px/inherit;;inherit;;inherit></font>
Para enviar este script a slurm, crear un job, y comenzar el procesamiento se requiere lo siguiente:
chmod +x ejemplo2.sh
sbatch ejemplo2.sh
Este ejemplo enviar un trabajo de 1 hilo a slurm con los parametros del script, y con limitaciones establecidas al usuario y la partición
4. Ejemplo básico 3 (Array Jobs)
Este ejemplo muestra como enviar varios tareas utilizando la propiedad de array-jobs en slurm, para más detalles consultar documentación oficial de slurm para ver todas las posibilidades ofrecidas en Array-Jobs.
#!/bin/bash
#SBATCH -J R-NOMBRE-SIMULACION #Nombre de la simulacion reemplazar segun el caso
#SBATCH -a 1-11%3 #
#SBATCH --nodes=1 #Número de nodos donde se enviara el trabajo, siempe es 1 para Soroban
#SBATCH --tasks-per-node=1 #Número de hilos a ejecutar simultaneamente
#SBATCH --mem=100G #Memoria reservada para la simulacion
#SBATCH --partition=intel #Particion general donde se enviara la simulación, llamada 'intel'
module load R/3.6.1 #Modulo necesario cargado previamente, necesario para la simulación
cmds=( #comandos a ejecutar en array
'sleep 10;echo 10'
'sleep 20;echo 20'
'sleep 30;echo 30'
'sleep 40;echo 40'
'sleep 50;echo 50'
)
eval ${cmds[$SLURM_ARRAY_TASK_ID - 1]} #desplegar informacion de los ID de cada elemento de array-job enviado
chmod +x ejemplo3.sh
sbatch ejemplo3.sh